Mol Cancer Ther. Author manuscript; available in PMC Mar 1, 2013.
Published in final edited form as:
Mol Cancer Ther. Mar 2012; 11(3): 526–537.
Published online Feb 17, 2012. doi: 10.1158/1535-7163.MCT-11-0806
PMCID: PMC3297694
NIHMSID: NIHMS345601
Copyright notice and Disclaimer
Novel therapies for metastatic renal cell carcinoma: Efforts to expand beyond the VEGF/mTOR signaling paradigm
Sumanta Kumar Pal, MD, Assistant Professor, Stephen Williams, MD, David Y. Josephson, MD, Courtney Carmichael, MSN, AOCNP, Nicholas J. Vogelzang, MD, Chair and Medical Director, and David I. Quinn, MBBS, PhD, FRACP, FACP, Associate Professor
Sumanta Kumar Pal, Division of Genitourinary Oncology, Co-Director, Kidney Cancer Program, Department of Medical Oncology & Experimental Therapeutics, City of Hope Comprehensive Cancer Center, Phone: (626) 256-4673, Fax: (626) 301-8233;
Contributor Information.
Sumanta Kumar Pal: spal@coh.org; Stephen Williams: williams@ocurology.com; David Y. Josephson: josephsond@towerurology.com; Courtney Carmichael: ccarmichael@coh.org; Nicholas J. Vogelzang: nicholas.vogelzang@usoncology.com; David I. Quinn: diquinn@usc.edu
Small right arrow pointing to: The publisher's final edited version of this article is available free at Mol Cancer Ther
Small right arrow pointing to: See other articles in PMC that cite the published article.
Other Sections▼
Abstract
With 6 agents approved for metastatic renal cell carcinoma (mRCC) within the past 5 years, there has undoubtedly been progress in treating this disease. However, the goal of cure remains elusive, and the agents nearest approval (ie axitinib and tivozanib) abide by the same paradigm as existing drugs (i.e., inhibition of vascular endothelial growth factor, VEGF, or mammalian target of rapamycin, mTOR, signaling). The current review will focus on investigational agents that diverge from this paradigm. Specifically, novel immunotherapeutic strategies will be discussed, including vaccine therapy, cytotoxic T-lymphocyte antigen 4 (CTLA4) blockade, and programmed death-1 (PD-1) inhibition, as well as novel approaches to angiogenesis inhibition, such as abrogation of Ang/Tie-2 signaling. Pharmacologic strategies to block other potentially relevant signaling pathways, such as fibroblast growth factor receptor (FGFR) or MET inhibition, are also in various stages of development. Although VEGF and mTOR inhibition have dramatically improved outcomes for patients with mRCC, a surge above the current plateau with these agents will likely require exploring new avenues.
Keywords: renal cell carcinoma, targeted therapy, XL184, CVX-060, AMG-386, BMS-936558, AV-951
Other Sections▼
Introduction
Without question, the treatment of metastatic renal cell carcinoma (mRCC) has evolved markedly in recent years. Prior to the introduction of targeted agents more recently, the mainstay of therapy were immune-directed agents such as interleukin-2 (IL-2) or interferon-a (IFN-α). Although IL-2 offers the potential for a durable remission in approximately 5–7% of patients, the vast majority obtain limited clinical benefit.(1) Similarly, the clinical efficacy of IFN-α is quite limited – meta-analytic data from the cytokine era suggests a median progression-free survival (PFS) of 4.7 months and a median overall survival (OS) of 13 months with this therapy.(2) In 2002, it was proposed that these values serve as a benchmark for future therapies for mRCC. Several targeted agents have now surpassed this benchmark. In a phase III study, the vascular endothelial growth factor-tyrosine kinase inhibitor (VEGF-TKI) sunitinib led to an improvement in OS as compared to IFN-α in treatment-naïve patients with clear cell mRCC.(3) In a phase III study enrolling poor-risk patients with mRCC, the mammalian target of rapamycin (mTOR) inhibitor temsirolimus similarly showed a survival advantage over IFN-α.(4) Data from these and other randomized trials have led to the approval of 6 agents in less than 5 years for advanced RCC. A limitation of this milestone, however, is that these agents share common molecular targets. Akin to sunitinib, the monoclonal antibody bevacizumab and the TKIs sorafenib and pazopanib target signaling via the VEGF receptor (VEGFR).(5–8) Also, similar to temsirolimus, everolimus is a small molecule inhibitor of mTOR.(9)
Because of this conundrum, the research community is at a crossroads. Should further research be directed at developing agents that also antagonize VEGF- or mTOR-mediated signaling? Over the past year, the VEGF-TKI axitinib met its primary endpoint in a phase III study, showing an improvement in PFS as compared to sorafenib in patients with mRCC refractory to first-line therapy.(10) Data for other VEGF-TKIs, such as tivozanib (AV-951), are eagerly anticipated.(11) At some point, however, it is possible that a ceiling effect may occur with these therapies. Experiences to date suggest that not all patients will obtain benefit from VEGF- or mTOR-directed treatments and, even amongst those that do, responses are unlikely maintained indefinitely. Thus, parallel efforts are in place to investigate novel signaling axes which may offer unique benefit to patients beyond existing therapies. Herein, these efforts will be described in detail.
Other Sections▼
Angiogenesis Inhibition: Unique Strategies Beyond VEGFR Targeting
Inhibition of the Ang/Tie-2 signaling axis
The majority of angiogenesis inhibitors used in clinical practice today function via direct inhibition of VEGFR. However, other putative approaches exist to disrupt tumor blood vessel growth and formation. Targeting Tie-2 signaling is one such strategy. The Tie-2 receptor is expressed principally on the vascular endothelium and knockout leads to embryonic lethality in murine models due to vascular disruption.(12) Signaling via Tie-2 is mediated by several key ligands, including angiopoietin-1 (Ang-1) and angiopoiein-2 (Ang-2). Ang-1 was been shown to bind to the Tie-2 receptor and up-regulate survivin, an inhibitor of apoptosis, in endothelial cells.(13) The net effect of this interaction is stabilization of vasculature. In contrast, binding of Ang-2 to Tie-2 leads to increased endothelial cell proliferation. In a rat corneal model of angiogenesis, Ang-2 blockade prevented VEGF-induced neovascularization.(14) Of interest, Ang-2 concentrations appear to be higher in patients with RCC, suggesting the potential role of this moiety as a therapeutic target. Tie-2 gene expression appears to correlate with Ang-2 expression in tumors, suggesting the potential role of both as putative targets. (15) In a correlative study including 34 patients with mRCC treated with standard doses of sunitinib, blood was collected at the start of therapy and during the course of treatment.(16) A total of 20 patients ultimately had progression on sunitinib therapy – in this subgroup, Ang-2 levels decreased in 18 patients (90%) after initiation of sunitinib, but increased in 14 patients (70%) at the time resistance was evoked.
Several clinical strategies have been employed to abrogate signaling through the Ang/Tie-2 signaling axis. The compound AMG-386 is a peptibody that disrupts the interaction of Ang-1 and Ang-2 with Tie-2. In a phase I clinical trial including 32 patients, the most commonly incurred toxicity was fatigue and peripheral edema.(17) Patients received weekly intravenous doses of AMG-386 at up to 30 mg/kg; of note, no maximum tolerated dose (MTD) was reached. Ten patients (32%) were noted to have some degree of radiographic shrinkage, although only one partial response was observed in a patient with refractory ovarian cancer. Four patients (13%) were noted to have stable disease (SD) for greater than 16 weeks. These results culminated in a randomized, phase II study exploring the agent in patients with mRCC.(18) In this study, patients were randomized in a 1:1:1 ratio to receive either sorafenib with AMG-386 at 10 mg/kg intravenous (IV) weekly (Arm A), sorafenib with AMG-386 at 3 mg/kg IV weekly (Arm B) or sorafenib with IV placebo weekly (Arm C). A total of 152 patients were randomized, with a PFS of 9.0, 7.5, and 9.0 months in Arms A, B and C, respectively. For the comparison of Arms A and B combined versus Arm C, the hazard ratio (HR) for PFS was 0.88 (95% CI 0.68–1.14, P=0.523). Although disappointing that the primary endpoint of improved PFS was not met, several items warrant mention. First, the observation of a 9.0 month PFS in association with sorafenib monotherapy is higher than expected based on the phase III experience leading to the approval of the drug, where a PFS of 5.5 months was observed. Second, the combination of sorafenib with AMG-386 did appear to have modest antitumor activity as compared to sorafenib alone. The maximum change in the sum of longest diameters (SLD) from baseline to post-baseline nadir was −34.3%, −29.2%, and −25.2% in Arms A, B, and C, respectively. Similarly, the response rate was higher in treatment arms containing AMG-386 (38%, 37%, and 24% in Arms A, B, and C, respectively). Added toxicity from AMG-386 appeared to be modest, with the safety profile for combination therapy resembling that of sorafenib monotherapy. Specifically, the most frequently incurred adverse events were diarrhea, hand-foot syndrome (HFS), alopecia and hypertension.
A second approach to abrogating Ang/Tie-2 signaling is selective targeting of the ligand. One such agent, CVX-060, is a fusion protein comprised of two Ang-2 binding peptides.(19) Preliminary results from a phase I study including 34 patients have been recently reported. With data available for 30 of these patients, no MTD was reached with doses escalating to 15 mg/kg IV weekly. The toxicity profile of the agent appeared to be relatively mild. Fatigue represented the most common adverse event, occurring in 23% of patients. Proteinuria (primarily grade 1/2) and hemorrhage (grade 1) were observed in a low percentage of patients (17% and 7%, respectively). A total of 24 patients (71%) remained on study therapy for ≥ 8 weeks. A randomized, phase II study will compare axitinib with or without CVX-060.(20) The study is anticipated to open in December of 2011, and will enroll a total of 165 patients. Following the theme of combining Ang-2 inhibition with VEGF inhibition, a distinct compound (CVX-241) is currently under development. Akin to CVX-060, this agent is a peptibody, but has affinity for both VEGF and Ang-2. Data from a phase I study evaluating CVX-241 has been recently reported. In 17 patients with solid tumors to date, no proteinuria or hemorrhage was reported (unlike the experience with CVX-060). The most commonly reported toxicities were fatigue, decreased appetite, back pain, and dyspnea. Of 13 evaluable patients, the best response observed is stable disease (SD) in 7 patients. It remains to be seen how the strategy of selective Ang-2 targeting will compare to the strategy of Tie-2 inhibition. Preclinical data from Coxon et al suggests that Ang-1 activity may be unmasked with use of Ang-2 inhibitors and thus dual inhibition of Ang-1/2 activity may be a preferred approach.(21)
Thalidomide and Lenalidomide
Despite widespread utilization of thalidomide and lenalidomide to treat multiple myeloma and myelodysplastic syndromes with 5q deletion, the mechanisms of these agents remain somewhat poorly understood. It is know that these agents have a complex effect on the tumor microenvironment, enhancing T-cell proliferation and modulating expression of various cytokines, including IL-2, IFN-γ, and IL-12.(22)
Escudier et al reported the activity of thalidomide in 40 patients with mRCC.(23) Patients received thalidomide at a starting dose of 400 mg daily, which was increased to 800 mg daily after 6 weeks if progressive disease (PD) was observed. If disease progression continued after a further 6 or 12 weeks of therapy at 800 mg dosing, the dose was then increased to 1200 mg. Of the 40 patients enrolled, 6 patients (15%) had received no prior therapy, while 8 (20%) had received 3 or more lines of treatment. Two patients (5%) exhibited a partial response (PR), and a median OS of 10 months was reported. The toxicities associated with thalidomide were substantial – for instance, of those patients who had received thalidomide for at least 12 months, 100% had demonstrable neuropathy on electromyography. A total of 9 patients (22.5%) developed thromboembolism, and 3 patients (7.5%) developed pulmonary embolism. Thus, the modest activity associated with thalidomide in mRCC was mitigated by the substantial toxicity profile.
Several other studies aimed to determine if lower doses of thalidomide could be combined with biologics. Hernberg et al assessed thalidomide at up to 300 mg daily with IFN-α dosed at 0.9 million international units (MIU) subcutaneously (SQ) 3 times daily.(24) With a total of 30 patients enrolled, 6 patients (20%) achieved a PR. Median time to treatment failure (TTF) was 7.7 months, and median OS was 14.9 months. When considering historical benchmarks, these results do not clearly suggest a benefit with the addition of thalidomide.(2) With this in mind, a phase III study comparing IFN-α with or without thalidomide may more definitively address this issue.(25) The study has been completed, although results have not yet been published. The combination of thalidomide and IL-2 has also been explored in 31 patients with mRCC. Patients received thalidomide at up to 400 mg daily in combination with IL-2 at 7 MIU/m2 with GM-CSF on days 1–5 from weeks 2 to 5 of therapy. After 7 weeks, patients repeated the same 6-week regimen up to 6 times. Clinical benefit was observed in 17 patients (55%), with 3 patients (10%) attaining a CR and 8 patients (26%) achieving a PR. The applicability of these results is limited by the IL-2 regimen utilized. Presumably, combination of high-dose IL-2 with thalidomide could result in substantially greater toxicity.
Phase II studies of lenalidomide have produced rather similar results. Chouieri et al reported results from a phase II study examining lenalidomide in 28 patients with mRCC who had received no more than 1 prior therapy.(26) Lenalidomide was administered at a dose of 25 mg daily for 3 weeks of a 4 week cycle. Three patients (15%) achieved a PR and remained progression free for longer than 15 months. A further 11 patients (39%) had stable disease lasting longer than 3 months, and median survival had not been achieved at the time of publication. The most frequent toxicities incurred with lenalidomide in this report were neutropenia, fatigue and dermatologic toxicity. A slightly larger experience was reported by Amato et al utilizing a similar schedule of lenalidomide in a total of 40 patients.(27) With 39 evaluable patients in this report, 1 patient achieved a CR and 3 patients (8%) achieved a PR. A further 21 patients (53%) were noted to have SD as a best response. Nine patients (23%) remained progression free after 12 months of therapy and a median OS of 17 months was reported. Akin to the experience reported by Chouieri et al, the most frequently incurred toxicities were neutropenia and fatigue.
Other Sections▼
Immunotherapy: Beyond IFN-α and IL-2
CTLA4 Inhibition
Pharmacologic blockade of CTLA4 prevents induction of T-cell anergy, which occurs when CTLA4 on the T-cell surface binds B7 on APCs.(28) Ipilimumab, a monoclonal antibody directed at CTLA4, has recently shown a survival benefit over gp100 vaccine in a phase III evaluation in advanced melanoma.(29) A phase II study was conducted in patients with clear cell mRCC utilizing two distinct dosing regimens, either (1) 3 mg/kg IV followed by 1 mg/kg IV every 3 weeks, or (2) 3 mg/kg IV every 3 weeks.(30) Of 21 evaluable patients treated at the lower dose, one patient had a PR. In contrast, 5 of 40 patients (12.5%) treated at the higher dose had a PR. Enteritis/colitis and dermatitis were the most common adverse events associated with therapy. Interestingly, those patients that developed autoimmune toxicities in association with ipilimumab therapy were noted to have a higher response rate (30%). Although there are no other active studies of ipilimumab in mRCC, a phase I study is currently assessing MDX-1106 in association with ipilimumab therapy in patients with stage III or IV melanoma.(31) If well tolerated, the regimen may be of interest in mRCC. It is unknown whether ipilimumab can be combined safely with current approved VEGF- or mTOR-directed therapies. However, a concerning signal has emerged from a phase I study assessing the CTLA4-directed monoclonal tremelimumab in combination with sunitinib in patients with mRCC.(32) Specifically, rapid acute onset renal failure was noted in a subset of 28 patients enrolled on this study. One patient receiving continuous sunitinib at 37.5 mg daily with tremelimumab at 10 mg/kg experienced sudden death, and 3 of 6 patients receiving the same dose of sunitinib in combination with tremelimumab at 15 mg/kg experienced DLTs.
Programmed Death-1 (PD-1) Inhibition
The interaction between PD-1 and its ligands, PD-L1 and PD-L2, play an integral role in regulating T-cell function. PD-1 is a transmembrane receptor on the T-cell surface, whereas its ligands are present on the surface of the antigen presenting cell (APC). The association of PD-1 and either PD-L1 or –L2 leads to induction of T-cell anergy. Thus, disrupting this interaction is a putative strategy to enhance the antitumor immune response. MDX-1106 is a fully human IgG4 antibody blocking PD-1. In a phase I study including 39 patients with either melanoma, colorectal cancer, castration-resistant prostate cancer, non-small cell lung cancer, or RCC, MDX-1106 was administered at doses of up to 10 mg/kg IV.(33) Three patients (7.7%) exhibited responses to therapy, including 1 complete response (CR) in a patient with colorectal cancer and 2 PRs in patients with melanoma and mRCC. Irrespective of dose, a sustained inhibition of PD-1 was observed, with persistent binding in over 70% of circulating T-cells ≥ 2 months following infusion. A separate phase Ib study sought to determine the safety and efficacy of MDX-1106 in a larger cohort of patients with a similar spectrum of malignancies.(34) Of 126 patients treated, 18 patients had mRCC and 16 of these patients had received MDX-1106 at the maximum dose (10 mg/kg). Patients received MDX-1106 for a median of 7.6 months, and 5 of 16 patients (31.2%) treated at the maximum dose achieved a PR. Six patients (37.5%) achieved SD as a best response. The most common toxicities incurred with therapy were fatigue, rash, pruritus and diarrhea, and one patient died of sepsis after developing grade 4 pneumonitis.
A randomized, phase II study is currently underway to further examine MDX-1106 in mRCC.(35) The study will allocate patients to one of three dose levels of the agent – either 0.3 mg/kg IV every 3 weeks, 2 mg/kg IV every 3 weeks, or 10 mg/kg IV every 3 weeks. A total of 150 patients who have progressed on at least 1 prior anti-angiogenic agent will be enrolled, and accrual is anticipated to complete by April of 2013. Further development of the drug may also include exploration of relevant therapeutic combinations, such as MDX-1106 in combination with currently approved VEGF-TKIs or mTOR inhibitors.
Vaccine Therapy
A multitude of vaccine-based approaches have been devised for the treatment of mRCC. The agent IMA901 was derived through a comprehensive analysis of multiple tumor specimens, primarily consisting of RCC. Tumor associated antigens (TAAs) were identified, including 9 HLA-class I and 1 HLA-class II binding peptides. These TAAs were noted to be highly immunogenic. In a phase II study, 68 patients with clear cell mRCC who had failed primary therapy with either cytokines or VEGF-TKIs were randomized to receive up to 17 vaccinations with IMA901 over a 9 month period with or without a single dose of cyclophosphamide at 300 mg/m2.(36) Survival at 12 months and 18 months was 67% and 54%, respectively. Disease control rate (DCR) at 6 months was higher in patients who had failed prior immunotherapy as compared to the post-TKI group (31% v 12%). With respect to the contribution of cytotoxic chemotherapy, it was noted that Treg quantity 3 days following treatment was markedly reduced in patients who received cyclophosphamide as compared to those who did not (P=0.032).(37) Of interest, survival was improved in those patients who generated detectable T-cell responses to IMA901 (P=0.019). Of 31 patients who generated a multipeptide response, survival at 12 and 18 months was 73% and 63%, respectively. Furthermore, in 8 patients who had received prior cyclophosphamide and had a multipeptide response, 100% of patients were alive at these intervals. A potential caveat of this finding is that more debilitated patients may demonstrate a greater degree of anergy, and would be anticipated to have a poorer outcome. Comparison of patient characteristics in groups stratified by T-cell response could be useful.
Given the apparent efficacy and scant toxicity associated with IMA901 (the most common adverse event was mild infusion reactions), a phase III study is underway to evaluate the agent. In this study, 330 patients with treatment-naïve clear cell mRCC will the randomized to receive either sunitinib alone or sunitinib with IMA901 vaccinations over the course of 4 months.(38) Akin to the previously noted phase II experience, patients receiving IMA901 will additionally receive a single dose of cyclophosphamide and adjunctive GM-CSF therapy. The study is anticipated to complete accrual by April of 2014.
Autologous dendritic cell vaccines have recently established a role in prostate cancer therapy, with the approval of sipuleucel-T for asymptomatic or minimally symptomatic castration resistant disease.(39) A slightly distinct approach has been taken in the domain of mRCC. AGS-003 represents an autogolous immunotherapy product derived from matured dendritic cells that have electroporated in the presence of tumor-derived RNA and CD40 ligand (the latter binds to CD40 on APCs and triggers activation). In a phase II study, AGS-003 was administered to 25 subjects with newly-diagnosed mRCC in association with sunitinib therapy. The vaccine was administered every 3 weeks for a total of 5 doses, and then every 3 months until PD was observed. Of note, no good-risk patients were included in the study – in the intention-to-treat (ITT) population (n=21), 15 patients had intermediate-risk disease while 6 patients had poor-risk disease. PFS in this collective group was 12.5 months. Notably, PFS appeared to be correlated with decreased regulatory T-cell function (r2=0.7662). In addition, patients with a prolonged PFS (i.e., exceeding 10 months) were noted to have expansion of CD27+ memory T-cells. A phase III study assessing sunitinib with or without concomitant vaccination with AGS-003 is anticipated.
Allogeneic vaccines are also under study for mRCC, albeit in a more preliminary phase. Fifteen patients were treated in a phase I study assessing administration of irradiated cells derived from a modified RCC-26 cell line.(40) The modified cell line had increased immunogenic potential via IL-2 secretion and expression of CD80 co stimulatory molecules. The vaccine was administered at doses of up to 40 × 106 cells over 22 weeks in patients with at least one metastatic site. Although no PRs were encountered, a median PFS of 5.3 months was observed. Median OS in the study was 15.6 months. Notably, patients with delayed-type hypersensitivity skin reactions to the vaccine demonstrated a longer survival in this initial report. A distinct allogeneic vaccine, MGN1601, has also been assessed in patients with mRCC. The vaccine is generated from human RCC cells that have been modified to express IL-7, GM-CSF, CD80, and CD154.(41) The vaccine also contains the TLR9-agonist dSLIM-30L1.(42) In murine studies, the vaccine greatly enhanced autoimmune responses, increasing infiltration of CD4, CD8, and CD86 cells up to 20-fold. Phase I/II testing of MGN1601 began in November of 2009, and clinical data associated with this agent is eagerly awaited.
Cytotoxic Therapy: A Resurrection?
Cytotoxic agents are still often employed as a salvage approach for patients with mRCC – most frequently, combinations of fluoropyrimidines with the nucleoside analogue gemcitabine are employed. In a phase II study, 41 patients were treated with continuous infusion 5-fluorouracil (5-FU) and gemcitabine.(43) Of these patients, 23 (57%) had received two or more prior regimens (either chemotherapy or immunotherapy). In this heavily pre-treated population, a modest response rate was observed amongst 39 evaluable patients – 7 patients (17%) achieved a PR, while 5 further patients had minor responses. Several permutations of this regimen, including capecitabine with gemcitabine with or without targeted agents, have been reported in multiple subsequent studies.(44–48)
Since the phase II data of 5-FU/gemcitabine was reported in 2000, several other cytotoxic regimens have been attempted. Most recently, the agent S-1 was examined in a phase II clinical study. S-1 represents an oral agent combining three components: (1) tegafur, (2) potassium oxonate, and (3) 5-choloro-2,4-dihydroxypyridine.(49) The benefit of S-1 over other oral fluropyrimidine formulations is derived from the fact that the two additional biologic modifiers may increase the antitumor activity and reduce bowel toxicity associated with tegafur.(50) The phase II experience enrolled 45 patients with mRCC who had received nephrectomy in addition to cytokine therapy (or, alternatively, patients who were cytokine ineligible). Anorexia and neutropenia were the most frequently encountered grade 3/4 adverse events, occurring in 8.9% of patients. A total of 11 patients (24.4%) demonstrated a PR, while an additional 28 patients (62.2%) had SD as a best response. Median PFS for the overall study population was 9.2 months, while the median OS had not been reached with a median follow-up period of 21.7 months. PFS was significantly longer in patients with low thymidylate synthetase (TS) mRNA expression (P=0.006). Furthermore, the TS mRNA levels were noted to be lower in responders to S-1 therapy (P−0.048).
With the difficulties of cross-trial comparisons in mind, at first glance, these results appear to be somewhat comparable to the results achieved with currently available VEGF- directed therapies in the treatment-naïve and cytokine-refractory setting. However, no phase III trials are currently underway to further evaluate S-1 in this setting.
Several studies have also attempted to define the efficacy of ixabepilone, a novel epothilone with activity in breast cancer, in the setting of mRCC.(51–53) In one phase II experience, patients with mRCC with any number of prior therapies were treated with ixabepilone at 40 mg/m2 IV every 21 days.(54) In the first 12 patients enrolled, no objective response were observed and the median time to progression (TTP) was only 2.3 months. The most common grade 3/4 toxicities encountered were lymphopenia, neutropenia, diarrhea and infection. Using a distinct schedule of ixabepilone in a separate phase II study, a far better efficacy profile was achieved, albeit in a less heavily pre-treated population. In this study, a total of 87 patients with mRCC who had not received prior chemotherapy or targeted therapy were treated with ixabepilone at a dose of 6 mg/m2 IV daily for 5 days every 3 weeks. One CR was observed, and 10 patients further demonstrated a PR as a best response, yielding an overall response rate (ORR) of 12.4%. A further 59 patients (67.8%) demonstrated SD as a best response and the median TTP for the overall study population was 4.8 months. To facilitate comparisons to contemporaneous publications of data related to sunitinib and sorafenib, the authors of this study further reported OS data for patients with clear cell histology and Motzer grade 0 or 1 disease. In this cohort of 74 patients, median OS was 19.3 months.
Targeting MET in mRCC
MET has a number of purported roles in the pathogenesis of RCC. Over a decade ago, germ line and somatic mutations were identified in the tyrosine kinase domain of MET in patients with papillary RCC.(55) MET may also play a critical role in clear cell RCC – inactivation of VHL may actually cause constitutive activation of the moiety, and VHL null RCC cell lines appear to be exquisitely sensitive to MET shRNA.(56–57) Tissue microarray (TMA) data incorporating 317 unique RCC specimens suggested higher expression of MET in tumor tissue relative to paired normal tissue across histologic subtypes.(58) Furthermore, increased MET expression was associated with increased tumor grade (P=0.0019), advanced clinical stage (P=0.021), and decreased survival (P=0.017). For these reasons, targeting MET may have relevance across RCC histologies.
The dual VEGFR2/MET targeting agent, XL184, has recently shown unprecedented activity in the setting of metastatic castration resistant prostate cancer (mCRPC), causing regression of metastases visualized on bone scan in 56 of 65 evaluable patients (86%) enrolled in a randomized, phase II study.(59) Early experiences with XL184 also indicate substantial activity in ovarian cancer and medullary thyroid carcinoma.(60–61) Preliminary results from a drug-drug interaction study assessing the combination of XL184 with rosiglitazone (a CYP2C8 substrate) also indicates impressive activity. Patients on the study had either differentiated thyroid cancer or mRCC with a clear cell component. Amongst 9 patients with mRCC, 4 patients (44.4%) demonstrated a PR – 7 of these patients had received ≥ 2 prior therapies. Given these promising preliminary results, the further development plan for XL184 in mRCC is eagerly anticipated.
ARQ197 is a highly-selective small molecule inhibitor of MET. The agent was recently assessed in a phase I study including 51 patients with advanced solid tumors.(62) Uniquely, the study incorporated paired biopsies performed prior to treatment and either at day 2 or 15 of therapy. Only one patient with mRCC was enrolled in this effort. SD lasting ≥ 4 months was the best response observed in the study, although minor tumor regressions were noted in gastric and Merkel cell tumors. With respect to the extensive correlative analyses performed in this study, marked reductions in total c-MET and phosphorylated FAK were observed. A phase II study of ARQ197 in microphthalmia transcription (MiT)-associated tumors offers a slightly larger experience with the agent in RCC. Amongst 28 patients enrolled at the time of a preliminary report were 4 patients with mRCC. Three of these patients (75%) achieved SD as a best response.(63) Tentative plans exist within the Southwest Oncology Group (SWOG) to assess the agent in patients with papillary mRCC (either with or without erlotinib).(64)
MET-driven tumor growth appears to be contingent upon ligand-activation by hepatocyte-growth factor (HGF).(65) In a series of 45 patients with previously untreated clear cell RCC, levels of HGF were higher as compared to non-cancer controls (P<0.0001).(66) Interestingly, in the subset of patients with higher Fuhrman grades and advanced stages, cause-specific survival was superior in those patients with higher levels of HGF. No such association was found with levels of VEGF.
HGF blockade has been examined as an antitumor strategy in mRCC. AMG-102 represents a monoclonal antibody with affinity for HGF.(67) In one phase II study, 61 patients with mRCC of varying histology and degrees of prior therapy were enrolled.(68–69) Although one patient incurred a confirmed PR that was maintained for over 2.5 years, SD was the best response in the majority of subjects (26 patients, or 43%). Grade 3/4 events occurred in 33% of the study population, with edema representing the most frequent adverse event. An assessment of baseline plasma levels of HGF and soluble c-MET was performed, although no correlation with efficacy was observed. Although clinical evaluation of AMG-102 is underway in a variety of other malignancies, it is unclear whether further assessment will proceed in mRCC.(70–71)
Targeting Fibroblast Growth Factor Receptor (FGFR) in mRCC
Emerging evidence suggests that FGFR may play a critical role in RCC pathogenesis. In 38 patients with mRCC, therapy with sunitinib was rendered and serial plasma collections were conducted during therapy.(72) In those patients who progressed, significant rises in bFGF levels were observed (P<0.01) – in contrast, no significant changes were observed in bFGF levels in those patients who exhibited responses or SD. Several other reports similarly suggest increased FGFR signaling as an escape mechanism for VEGF antagonism.(73–74) Dovitinib (TKI-258) represents a small molecule inhibitor with affinity for FGFR1-3.(75) Preliminary phase II results are available from a phase I/II evaluation of dovitinib in patients with mRCC. In 51 patients evaluable for efficacy, 4 patients (8%) demonstrated a PR, while 19 patients (37%) had SD ≥ 4 months as a best response. Notably, 3 patients who obtained a PR had prior therapy with both VEGF- and mTOR-directed therapies. Median PFS and OS in the study population overall was 6.1 and 16 months, respectively. In 59 patients evaluable for safety, the most common adverse events were nausea, diarrhea and vomiting, although grade 3/4 events occurred at a relatively low rate for each of these toxicities (< 10%). Studies of dovitinib are ongoing in a variety of other malignancies, including breast, gastric, and urothelial carcinoma, and a phase III study is underway comparing dovitinib and sorafenib in patients with mRCC who have failed prior therapy with mTOR-and VEGF-directed therapy.(76–79) The study is anticipated to enroll a total of 550 patients by May of 2013, and will assess a primary endpoint of PFS. Investigations of distinct FGFR inhibitors (e.g., E-3810, brivanib, AZD4547, etc.) are also occurring simultaneously.(80–82) Amongst these agents, brivanib (a dual VEGFR/FGFR inhibitor) is being examined in a phase II trial in patients with clear cell mRCC that have progressed on prior VEGF-directed therapy.(83) Tumor assessments via 124I-cG250 PET/CT will be conducted in association with standard radiographic assessments in this study.(81)
Conclusions
Thus far, drug development in mRCC has followed a relatively predictable paradigm, with a steady stream of VEGF- and mTOR-directed therapies. A number of VEGF-antagonists remain in the pipeline, including axitinib and tivozanib.(84–89) Predicated on greater specificity and higher affinity for VEGF receptors, the clinical benefit of these agents over existing drugs appears to incremental at best. Multiple studies assessing the combination of VEGF- and mTOR-directed therapies are also underway.(90–92) Several combinations (i.e., bevacizumab with sunitinib) have been marred by substantial toxicity.(93) Early results from trials examining better tolerated regimens (i.e., bevacizumab with temsirolimus) appear to show little added efficacy.(94–96)
It therefore appears that substantial progress in the treatment of mRCC will be made by expanding beyond the existing paradigm and exploring novel pathways and therapeutic approaches. Several agents under investigation act on distinct moieties along the VEGF-/mTOR-signaling axis. For example, BEZ235 is a dual PI3K-/mTOR-inhibitor currently being examined in solid tumors – the agent appears to have activity in preclinical models of RCC.(97–99) BKM120 is a distinct PI3K inhibitor that is soon to be examined in a phase I study in combination with bevacizumab in patients with mRCC.(100–102) Data from several studies investigating the agent perifosine, a synthetic alkylphospholipid that modulates a number of signal transduction pathways including Akt, have indicated modest activity in patients refractor to both VEGFR and mTOR inhibitors.(103–104) Although clinical data for these agents are eagerly awaited, landmark improvements in clinical outcome for mRCC will more likely come from targeting entirely distinct pathways. Early efforts to do so have been fraught with challenges – ErbB-directed therapies (lapatinib and erlotinib), IL-6 targeting agents (CNTO-328), and thrombospondin-1 agonists (ABT-510) have yielded modest efficacy at best in the setting of mRCC, and have unclear development plans within the disease as a consequence.(105–107) However, many of the agents reviewed herein (vaccine therapies, PD-1 inhibitors, etc.) are based on an evolving understanding of RCC biology. While the goal of cure remains elusive, trials examining these agents represent a critical step forward.
​ |
 肺部进展,脑膜稳定,单倍阿美20个月
我母亲23年4月确诊肺腺癌即脑膜转,鞘注了几次后单倍吃阿美20个月了,最近检查肺部略
肺部进展,脑膜稳定,单倍阿美20个月
我母亲23年4月确诊肺腺癌即脑膜转,鞘注了几次后单倍吃阿美20个月了,最近检查肺部略
 HER2突变肺癌诊疗实现新突破,HER2检
作者:seacat
近几年来靶向和免疫疗法的发展显著改善了肺癌患者的生存期,但仍有一部
HER2突变肺癌诊疗实现新突破,HER2检
作者:seacat
近几年来靶向和免疫疗法的发展显著改善了肺癌患者的生存期,但仍有一部
 父亲晚期肺癌即将迈入第7年,这是我
讲述者:郭先生整理者:pear
2018年4月底,父亲因长期右肩胛骨疼痛、脸部及颈部肿大而
父亲晚期肺癌即将迈入第7年,这是我
讲述者:郭先生整理者:pear
2018年4月底,父亲因长期右肩胛骨疼痛、脸部及颈部肿大而
 疾病控制率89%!小细胞肺癌又有新靶
作者:seacat
B7-H3是近年热门的新兴靶点,有多种类型药物投入临床。目前实体瘤领域进
疾病控制率89%!小细胞肺癌又有新靶
作者:seacat
B7-H3是近年热门的新兴靶点,有多种类型药物投入临床。目前实体瘤领域进
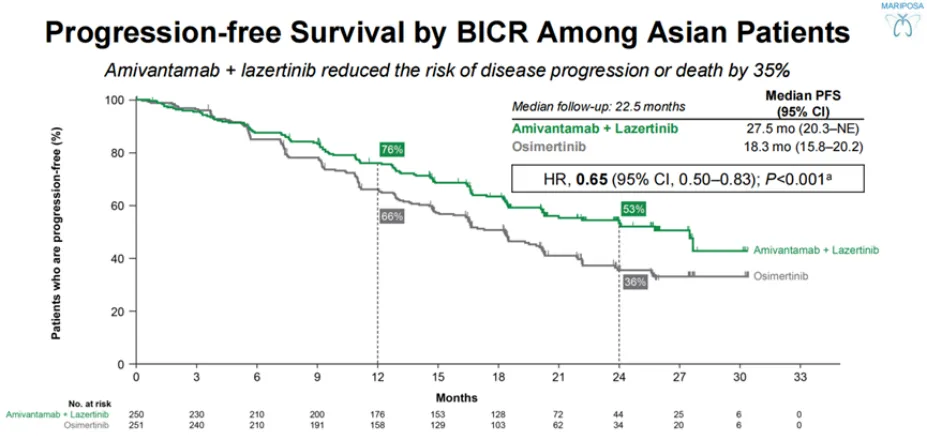 首个击败奥希替尼的药物组合获批!显
作者:seacat
2024年8月20日,强生宣布美国FDA批准埃万妥单抗(Amivantamab)+兰泽替尼
首个击败奥希替尼的药物组合获批!显
作者:seacat
2024年8月20日,强生宣布美国FDA批准埃万妥单抗(Amivantamab)+兰泽替尼




 显身卡
显身卡

